There has been a 60 year long discussion on the role of the neurotransmitter serotonin in the pathophysiology of depression. A recent systematic investigation by Joanna Moncrieff and colleagues concluded that “main areas of serotonin research provide no consistent evidence of there being an association between serotonin and depression, and no support for the hypothesis that depression is caused by lowered serotonin activity or concentrations”.
Yesterday, a new paper came out in which the authors made the strong claim that they found “clear evidence” for the serotonin theory of depression, that is, that the neurotransmitter serotonin is involved in the pathophysiology of depression.
Less than 24 hours after the paper appeared online, there has already been substantial media coverage, such as a piece in the Guardian. Given the relevance of the study, I’ll explain in this blog post why the paper’s findings do not support the conclusion the authors draw.
The study
The study, entitled “BRAIN SEROTONIN RELEASE IS REDUCED IN PATIENTS WITH DEPRESSION: A [11C]Cimbi-36 PET STUDY WITH A D-AMPHETAMINE CHALLENGE”, was published on November 4th 2022 in Biological Psychiatry. The Guardian summarizes the study well:
The participants were given a PET scan that uses a radioactive tracer to reveal how much serotonin was binding to certain receptors in the brain. They were then given a dose of amphetamine, which stimulates serotonin release, and scanned again. A reduced serotonin response was seen in the depressed patients, the researchers found.
In the core summary of their paper, the researchers conclude that the study “provides clear evidence for dysfunctional serotonergic neurotransmission in depression”——a strong claim. I show in this blog that this conclusion is absolutely not warranted given the presented evidence.
Sample size is small, generalizability is not given
The study involved 17 depressed participants and 20 healthy controls. I want you to keep in mind that the authors here wrote a paper about depression—they wanted to learn about depression, not about the 17 participants in particular. This is the main reason we use statistics in science: you study a small sample of interest, and then use statistics to draw inferences about the population you are interested in.
No matter if you have training in statistics or not, you likely have built a pretty good statistical intuition by reading results of election polls. Suppose I want to know which of 2 parties will win the next election in the Netherlands, and to do so I carry out a poll with 37 participants. You would be very skeptical when I tell you that there is “clear evidence” that party 1 will win over party 2 from this small sample. Of course you would be more confident if out of 37 participants, every single one said “party 1” and nobody said “party 2”. But even in this case, the problem that remains is generalizability: who these 37 participants are.
Suppose I told you that I recruited the 37 participants as randomly as I could, by traveling the Netherlands for weeks and asking every 1000th person I encountered. You would have more confidence in my results than when I told you all 37 participants I asked were asked during a campaign event of party 1.
Overall, sample size and generalizability are the reason why firm conclusions about e.g. “depression” only follow when 1) we draw a large sample from the population we are interested in, and 2) we draw a random sample of depressed patients.
In this particular study, neither is the case. The study has a very small sample size, and generalizability is very low because the depressed group is not representative of people with depression broadly. There are many factors here, but just to list one: 5 of the 17 depressed participants in the study (i.e. 30%) have Parkinsons disease, but this does of course not apply to the population of interest (not every third person with depression has also Parkinsons).
Even if the study had strong statistical results, the study cannot provide “clear evidence” for the role of serotonin in depression given these limitations. Unfortunately, the study does not have strong statistical results either.
There is little to no statistical evidence at all
The most important thing to know is that the authors themselves do not find a significant difference between depressed and healthy people regarding serotonin release. They do find a significant difference only after removing one of the 17 depressed participants. I will not go into the weeds of discussing in detail here whether this exclusion is warranted or not, because it does not matter: you do not have “clear evidence” for the serotonin theory of depression if your result depends on one particular participant in your study.
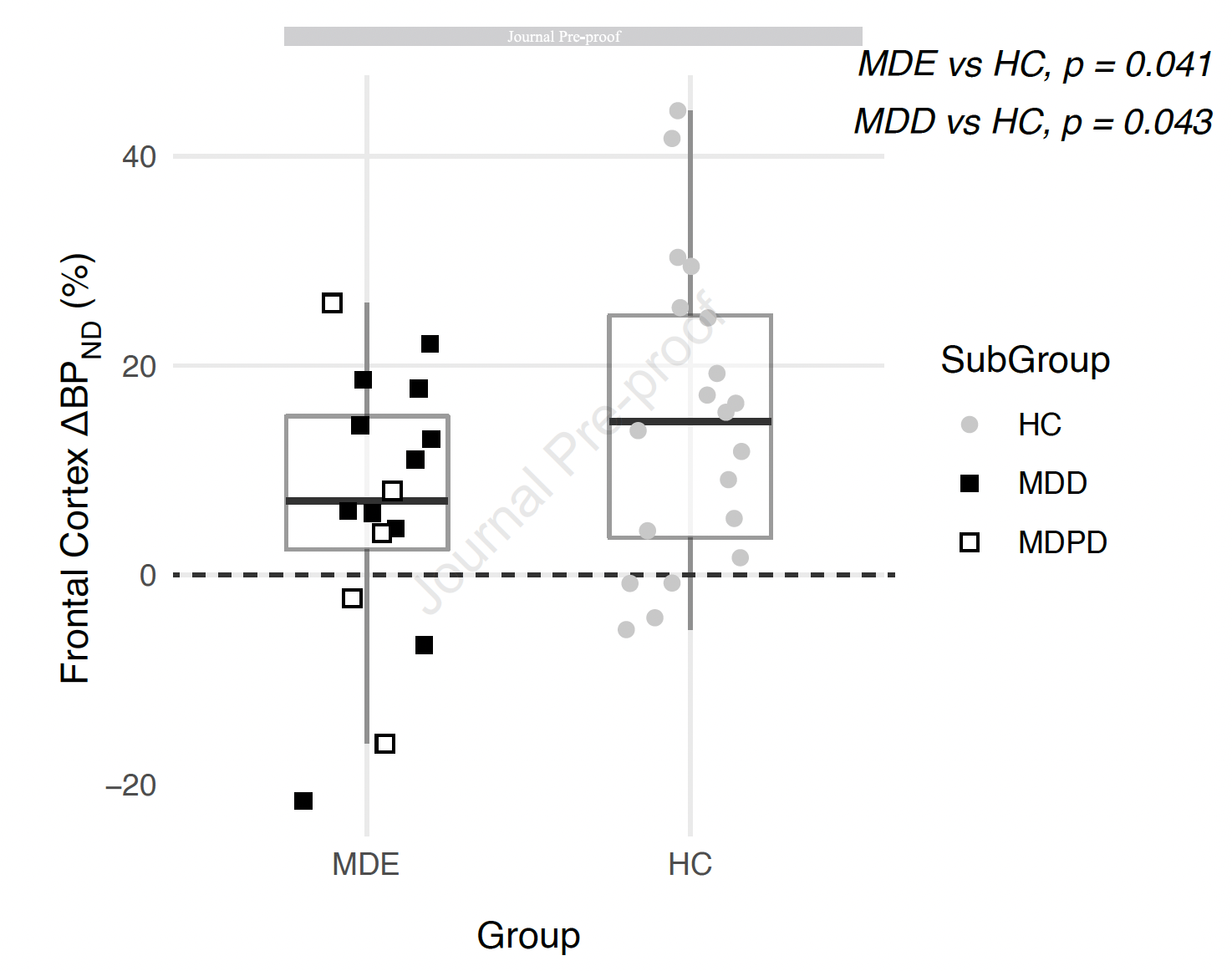
But even after removing this one person, results are anything but conclusive—see below. On the left (black dots), you see the depressed participants, on the right (grey dots) the healthy controls, and the y axis denotes serotonin release (the people with Parkinson were excluded from the plot, if interested you can find the plot with these people included here).
{kind=link}

Ron Pat-El on Twitter made an important point by marking up the plot:

As you can see, depressed people and healthy controls are nearly perfectly similar in their serotonin release, except for 1 depressed person (left bottom circle) and 2 healthy people (top right circle). This does not establish “clear evidence” for group differences. Consider the above plot again, and think of the y-axis as height of people rather than serotonin release. Based on the data points, you would not draw draw the conclusion that there is “clear evidence” for height differences (depressed people being smaller than healthy controls) given the evidence presented. Yes, there is one depressed person who seems quite small, and 2 healthy controls who are quite tall, but overall the two groups are very similar in overall height.
The authors conduct 3 statistical tests that compare the two groups of people regarding their serotonin release. Two of these tests are barely statistically significant (p-values are both 0.04, anything under 0.05 is considered statistically significant), and the third test is not significant. I want to stress here that statistically speaking, this is the weakest of evidence possible that still counts as a significant finding. There are many ways a scientist can test whether something is significant or meaningful or interesting. If you use a p-value, the threshold for significance the authors choose here is 0.05, but it is also common to choose much more stringent thresholds, such as 0.01 or even 0.001. According to these rules, the finding would not be significant. One can also estimate a bayes factor, which compares competing hypotheses (“depressed people have lower serotonin release than healthy controls” vs “there is no difference”) and quantifies the support for one model over the other. If you calculate the bayes factor in this case, which the authors did not do, it is smaller than 1—this means that there is no support at all for the serotonin hypothesis of depression. A support for the serotonin hypothesis would start with a bayes factor larger than 1), and to conclude that there is “clear evidence”, as the authors do, would require a bayes factor of 5 or even 10.
The authors do not look at these ways to quantify evidence. They also do not calculate any effect sizes for their results. Effect sizes are different from p-values and tell us, independent from statistical significance, how different the two groups are we compare here in terms of magnitude of serotonin release. This is commonly done in statistics, and I believe authors did not report effect sizes because the magnitude of difference here is really minimal. And when there is a minimal difference between healthy and depressed folks, it cannot corroborate theories about the pathophysiology about a disorder.
Another way of thinking of the magnitude of effects is by dropping random participants. Erick Turner uses the metaphor of a missed taxi, which I really like: imagine you claim that your study provides “clear evidence” to support a theory. Now imagine that the one depressed participant in the very bottom left of the plot, the outlier, misses their taxi on the day of the study and cannot join the investigation. The results would no longer be significant, and there would not be any evidence to support the serotonin theory for depression from this study. Clearly, your result is not robust if a single person participating in your study or not changes your results dramatically. For ”clear evidence”, you need a much larger samples in which such individual outliers do not impact on core findings. Think back to the poll we discussed above: if one person less in a poll changes your result from “clear evidence that party 1 wins” to “clear evidence that party 2 wins”, you would disregard it as uninformative.
In addition to testing whether there is more serotonin released in response to amphetamines in the brains of depressed people vs healthy people, the authors also investigate whether depression severity (rather than depression diagnosis) is associated with serotonin levels (or changes of serotonin levels; both questions were investigated). Finding a significant relation here is important to support the idea that serotonin hypothesis of depression, which maintains that lower serotonin levels in depressed people play a causal role in the pathophysiology of depression. Accordingly, the authors hypothesized in the paper that there would be a relationship between serotonin levels and depression severity (the lower the serotonin level, the higher the depression severity). The authors found no statistical association, concluding in the paper that “at this stage we have no explanation for the lack of such relationship.” If you don’t find a relationship between serotonin levels and depression severity, how can you confidently conclude that there is “clear evidence” in support of the serotonin hypothesis for depression?
Conflicts of interest
Finally, I’d like to note that the conflict of interest statement seems incorrect, which states that “all authors report no biomedical financial interests or potential conflicts of interest”. Without having the time to dig into this deeper for all authors, I know that Dr Nutt has previously declared COIs related to biomedical research, e.g. “D.N. received consulting fees from Algernon and H. Lundbeck and Beckley Psytech, advisory board fees from COMPASS Pathways and lecture fees from Takeda and Otsuka and Janssen plus owns stock in Alcarelle, Awakn and Psyched Wellness” in a study published just a few months ago.
But this can still be fixed, given that the paper on the website of the publisher is currently the “pre-proof” version of the paper. This likely also explains a number other inaccuracies or inconsistencies I found in the paper, such as a p-value for the main finding of 0.038 in the abstract of the paper that does not appear in the results (instead, the authors report 0.041 lateron).
Conclusion
I want to address one common point folks have brought up in response to criticism of the paper yesterday, e.g. on Twitter, to defend the work: “PET studies are hard to carry out, and it is expensive and difficult to recruit large samples”. This may be true, but it has nothing to do with the arguments critics raised.
As I explain in great detail elsewhere, small samples are not inherently problematic, and it is perfectly fine to have a sample size of 1 participant. The problem is when researchers draw conclusions from studies that do not follow from the presented evidence, for example, because of validity threats due to the fact that the sample is too small. In an n=1 study, or this study with 17 depressed participants, it’s perfectly fine to argue that one learned something about one person, or 17 people respectively. And one can even establishing interesting preliminary findings if results are strong enough, and call for replication studies in larger samples. But one cannot learn much about “depression” from studying 17 people with depression—threats to external validity, and the heterogeneous nature of depression stand in the way of such conclusions.
As Hannah Devlin quotes me in the Guardian piece: “The conclusions the authors draw are not proportional to the evidence presented”, and that is the problem, not the sample size.
There are many other issues I see with the paper I won’t discuss in detail here, but one is worth mentioning, perhaps: since the serotonin theory of depression became popular in the 60s, we have learned a lot about depression that just doesn’t align with the theory. One of these insights is that the diagnosis of major depressive disorder places people who have very diverse problems and etiologies into one single group that hinders effective treatments, and that most one-size-fits-all treatments for depression haven’t worked out so well.
Few researchers today still believe that it is plausible that there is one particular causal (biological, psychological, or social) mechanisms such as serotonin that would be the main driver of depression; to remind you, this is exactly what the serotonin theory of depression claims (folks in the 60s often talked about a “final common biological pathway” for depression). Instead, as we have shown in two recent papers published in the last few months, depression is probably best conceptualized as emerging from a system of biopsychosocial components that interact with each other in complex ways. The role of serotonin in this system deserves further consideration. Here is a brief blurb from our recent work on systems; the two full papers can be found here (Nature Reviews Psychology) and here (Current Directions in Psychological Science).
In the summer of 2019, a scholar I greatly admire was kind enough to lend me his bicycle for a few months, granted I take good care of it. When the bicycle broke down after 3 weeks, I was terribly worried, but reductionism came to the rescue: Bikes can be decomposed into their constituent parts, and fixing all parts at the micro level will restore function at the macro level. But mental disorders are not like bicycles—they are like many other complex systems in nature. Whether a lake is clean or turbid results from interactions of interdependent elements, such as oxygen levels, sunlight exposure, fish, pollution, and so on. Whether my mood while writing this manuscript is anxious or cheerful is the outcome of causal relations among elements of my mood system, including my personality and disposition; the previous night’s sleep; my caffeine consumption; and external influences such as my email inbox. The same applies to mental health states. From a systems perspective, such states result from interactions of numerous biological, psychological, and social features, including specific risk and protective factors, moods, thoughts, behaviors, biological predispositions, and social environments.
UPDATE 1, November 13 2022: I just saw that the Guardian has now changed their original headline from “Study finds first direct evidence” to “Study claims to find first direct evidence” a few days after my blog went live. And it turns out I wasn’t the only one who thought the conclusions draw in the study don’t follow from the evidence.

UPDATE 2, June 25 2023: There is now a highly critical commentary on the Moncrieff et al. paper, along with a rebuttal by the original authors.
Thank you for sharing your critical insights and keeping us up to date of this discussion.
Pingback: “Clear evidence” for serotonin hypothesis of depression? – A New Vision for Mental Health